
怎么编写:欧盟新版CE临床评价(MEDDEV 2.7.1 Rev 4)
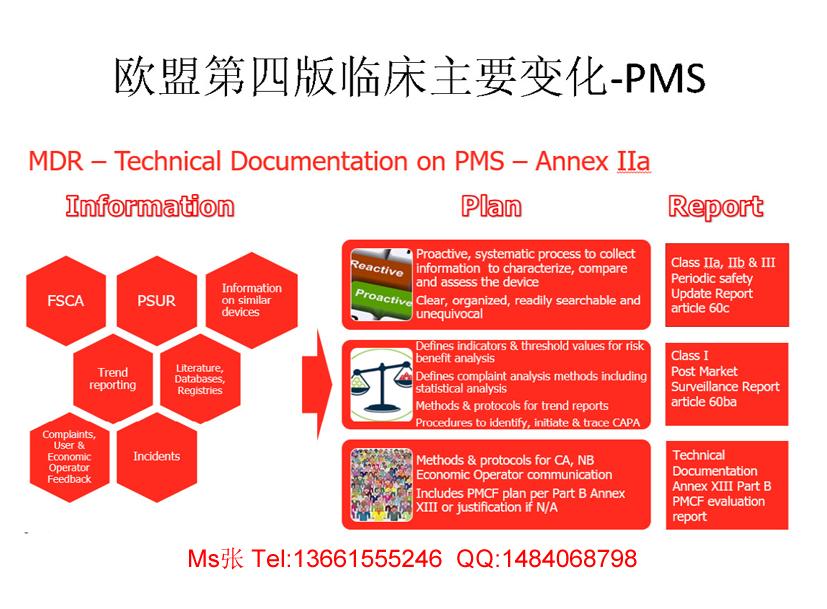
2016年6月发布的***器械临床评价指南MEDDEV2.7.1第4版替代了2009年12月发布的第3版。不仅第4版的长度从第3版的46页增加到了65页,而且第4版还包含了对临床数据更详细更广泛的要求。***器械出口企业在申请CE认证时,不管是I类普通产品还是II/III类高风险产品,都必须要提供第四版临床报告。并且已经拿到CE证书的企业,今年监督审核也需要提供。该版本的要求针对于MDD指令和AIMD指令,所以,将要申请或者已经拿到了TUV莱茵、TUV南德、SGS或其他公告机构CE证书的企业,一定要高度关注。这篇文章通过回答以下6个问题来帮助您了解新版的MEDDEV2.7.1:更多信息,随时咨询小张1366-1555-246***1484-0687-981.不变的是什么?变化的是什么?一般原则仍然是制造商必须使用临床数据来证明器械符合相关的基本要求(EssentialRequirements)。这些临床数据仍须基于本器械的(上市前)研究数据、其他同类器械的研究数据以及来自上市后监督(PMS)活动和警戒活动的数据。仍需对临床数据进行收集、评价和分析,但是,临床评价应该详尽到什么程度、什么评价方法是可接受的以及应该何时进行临床评价,这些内容几乎是全新的。2.何时进行临床评价?临床评价贯穿于***器械的整个生命周期,包括器械的设计阶段。临床评价是一个持续不断的过程,应当有相应的文件记录。对于临床评价报告(CER)的更新频率,制造商应给出合理解释,更新频率应根据风险、科学发展、设计变更等相关因素来确定。若通过上市后监督(PMS)收集到可能改变临床评价的相关数据,应对临床评价报告进行修订。对于创新或高风险器械,其临床评价报告应每年更新一次。对于其它器械,其临床评价报告应至少每2到5年更新一次。制造商必须对更新频率做出合理解释。3.如何进行临床评价?临床评价的不同阶段涉及范围和计划的定义、数据识别和每个数据集的评价、数据分析、和临床评价报告的定稿。对于大多数制造商来说,临床评价并不陌生,不过新版MEDDEV2.7.1对这些阶段进行了详细的描述。新版指南也详述了进行临床评价的人员资质要求。另外,对于评价的范围,新版MEDDEV2.7.1提供了更详细的要求。同时新版指南也详述了在哪里进行文献检索、如何进行文献检索、以及如何记录这些文献的收集、评价和分析过程。4.是否仍可使用实质等同器械?新版指南下,仍可使用实质等同器械的数据。但是“实质等同”的概念在新版指南中有了明确的定义,解读的空间相比之前要小很多。实质等同器械应当几乎是完全相同的。新版MEDDEV2.7.1的附录A1对实质等同性证明进行了详细说明。要证明器械的实质等同,必须考虑到器械的临床、技术和生物特性。新版MEDDEV2.7.1的附录A1对这三方面的特性进行了详细说明。而且器械需要满足所有这三方面的特性才能证明实质等同。在此背景下,新版指南还描述了“差异”的概念,以及验证此概念所需的步骤。实际上,实质等同器械的使用将局限于同一制造商的器械,而且符合要求的器械必须来自相同的器械系列。即便是同一器械的换代产品,要想证明实质等同,也需仔细考量。5.临床评价报告(CER)长啥样?新版MEDDEV2.7.1的要求非常清晰,因为它不仅要求对临床数据进行分析,而且还要求制造商透明公开所采用的方法和步骤。临床评价报告的实质部分应包括一个日志,记录该临床评价是如何进行的。相应的,临床评价报告就必须有附录,包括检索策略、全部检索结果、评价策略和结果、数据分析、以及清晰的参考文献列表。此外,所有文章和报告应可供审核员核实之用。6.新的欧盟***器械***(MDR)情况如何?提议的***器械***(MDR)的妥协统一文本已于2016年6月发布。预计在2019年的***季度,新的***器械***(MDR)将正式实施。新的***器械***(MDR)要求高质量的临床评价。新版MEDDEV2.7.1向此方向迈出了坚实的一步。按照新版MEDDEV2.7.1进行临床评价也可帮助制造商为即将出台的立法做准备。关于MEDD***.7.1Rev4,我司可以协助您:1、协助建立临床评价程序;2、建立临床评价方案3、寻找等同产品,进行等同分析;4、搜索文献及其他临床数据;5、临床数据分析;6、完成临床评价报告。)